Alternative Splicing Analysis
 0
0
Posted: Tue July 16 11:15 PM PDT
Member: kiko Garcia
Tags: biotech
Overview
Alternative splicing is a complex cellular mechanism that is critical for proteome diversification and plays an important role in cellular development, function, and disease mechanisms. Given the importance of alternative splicing events, their accurate mapping and quantification are critical for downstream analysis, especially for linking disease to alternative splicing. With rapid advances in sequencing technology, tools tailored to specific sequencing methods are now available to delve deeper into the nuances of splicing patterns. Traditional short-read RNA-Seq methods are often unable to span full-length isoforms and require computational reassembly of them, which can lead to erroneous reconstructions. With long-read sequencing technologies, isoforms can be sequenced end-to-end in a single read, allowing them to be unambiguously characterized and simultaneously quantified in a single dataset.
What is Alternative Splicing?
Alternative splicing is a regulated cellular mechanism by which multiple mature mRNA molecules can be generated from a single pre-mRNA molecule. This complex post-transcriptional process ensures that distinct protein isoforms are produced from a single gene, thereby increasing the diversity and complexity of the proteome. The human genome contains approximately 20,000 protein-coding genes that can produce a large number of protein products through alternative splicing.
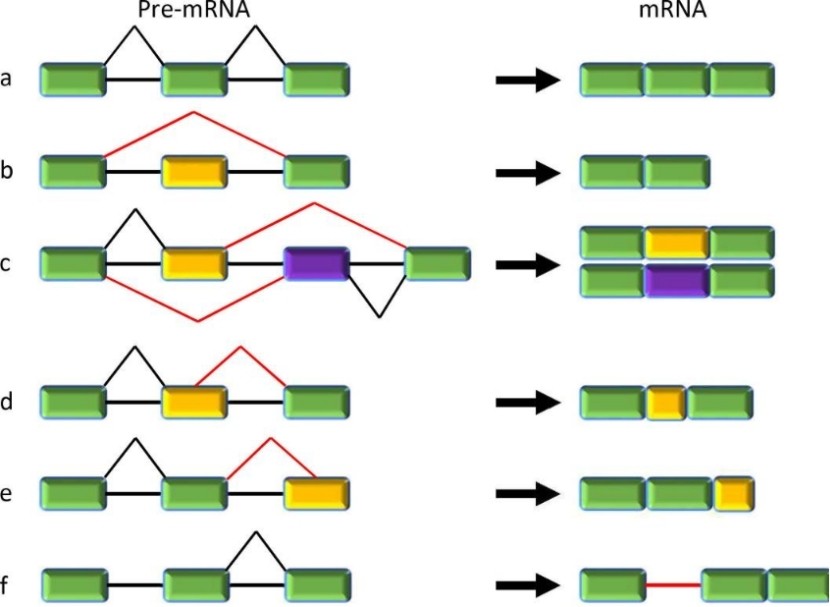
Constitutive and five major types of alternative splicing. (Jiang, et al., 2021)
Mechanism and Regulation of Alternative Splicing
The Complex Mechanism of the Spliceosome
At the heart of the alternative splicing mechanism is the spliceosome, a dynamic macromolecular machine. The spliceosome consists of five small nuclear ribonucleoproteins (snRNPs) and many associated proteins whose main role is to recognize and interact with specific sequences on pre-mRNAs. Notably, these include 5' and 3' splice sites, branch points, and polypyrimidine tracts. Through a complex series of molecular rearrangements, intronic regions are excised and exonic regions are joined together to produce mature mRNA.
Regulation by Splicing Factors
The decision matrix of which exons to include or exclude in the final mRNA is not random. It is orchestrated by splicing factors that either promote or inhibit the action of the splicing machinery. Key players in this regulatory dance include serine/arginine-rich (SR) proteins and heteronuclear ribonucleoproteins (hnRNP).
SR proteins: Usually act as splicing activators. They bind to exonic splicing enhancers (ESEs) and promote spliceosome recruitment to adjacent splice sites.
hnRNP: Often acts as a splicing repressor. Their association with exonic or intronic splicing silencers (ESS or ISS) prevents spliceosome assembly at adjacent splice junctions.
Environmental cues, cellular stress, or developmental stage may cause changes in the levels or activities of these splicing factors, which dictate changes in splicing patterns to suit the cellular environment.
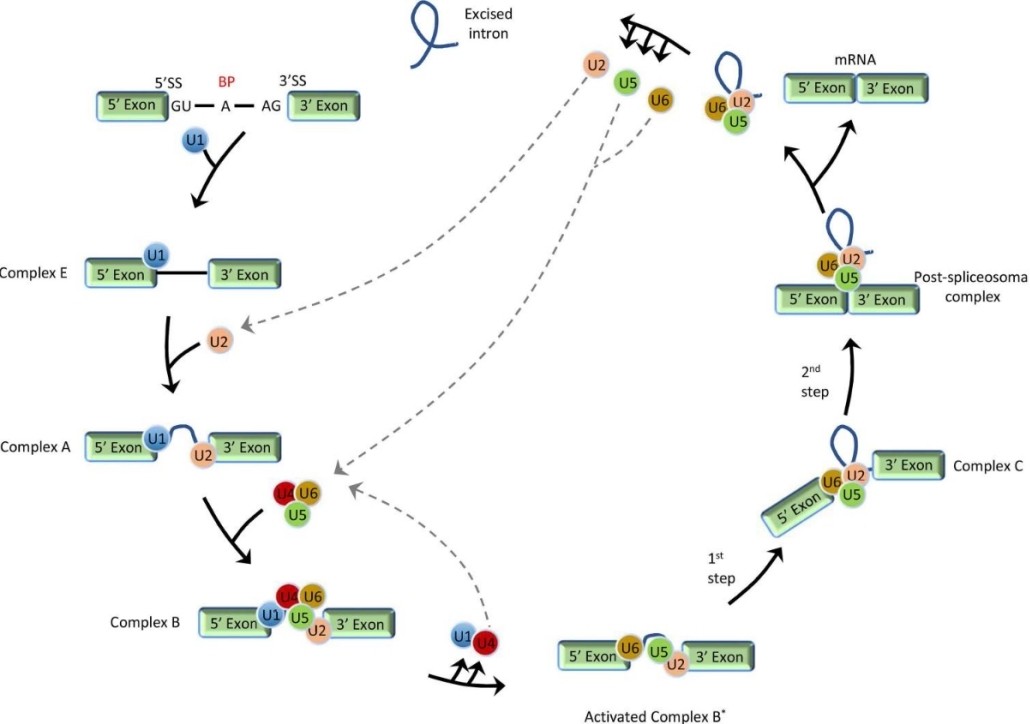
Stepwise schematic presentation of general pre-mRNA splicing. (Jiang, et al., 2021)
Human Diseases Associated with Alternative Splicing
Perturbations in the intricate molecular machinery governing alternative splicing can precipitate a spectrum of pathological states. Splicing aberrations, stemming either from mutations directly impinging on splice sites or dysregulation of auxiliary regulatory sequences, are intrinsically linked with a myriad of human pathologies.
Spinal Muscular Atrophy (SMA)
SMA exemplifies the profound consequences of splicing perturbations. The etiological underpinnings of this debilitating neurodegenerative disorder trace back to mutations in the SMN1 gene. However, the clinical trajectory and severity of SMA are modulated by alternative splicing events within its paralog, SMN2. Intriguingly, a mere single nucleotide polymorphism distinguishes SMN1 and SMN2, a discrepancy leading to the differential inclusion of pivotal exons in SMN2. This, in turn, culminates in a truncated protein variant with compromised functionality.
Cancer
The protean nature of alternative splicing is evident in its pervasive role in oncogenesis, shaping tumor biology, aggressiveness, and therapeutic susceptibility. The Bcl-x gene, pivotal in the apoptotic cascade, underscores this with its alternative splicing events birthing two isoforms: the anti-apoptotic Bcl-xL and its pro-apoptotic counterpart, Bcl-xS. A preponderance of Bcl-xL isoforms, resulting from splicing dysregulation, emerges as a recurrent theme across multiple malignancies, invariably fostering tumor resilience and chemoresistance.
Nervous system Disease
The neural milieu isn't immune to splicing aberrations either. A spectrum of neurological afflictions, encompassing both frontotemporal dementia and Parkinson's disease, betray splicing irregularities. Specifically, variations in the splicing patterns of the MAPT gene, encoding tau protein, have been implicated in a cluster of neurodegenerative tauopathies.
Cardiovascular Diseases
The intricate tapestry of cardiac physiology intertwines with alternative splicing. The cardiac troponin T (cTNT) gene, an orchestrator of cardiac muscle contraction, is susceptible to alternative splicing events. Erroneous splicing episodes in cTNT are pathognomonic of dilated cardiomyopathy, exerting detrimental effects on cardiac hemodynamics.
Methods for Characterizing Alternative Splicing
RT-PCR and qRT-PCR
Reverse transcription PCR (RT-PCR) and quantitative RT-PCR (qRT-PCR) are well-established methods for validating alternative splicing events identified by high-throughput methods. They can be used to quantify the expression of different splice variants and confirm the presence of specific splice junctions. These methods require careful PCR primer design to ensure specific detection of specific spliced isoforms.
RNA-seq has revolutionized transcriptomics by providing a high-throughput approach to the study of transcripts. In the context of alternative splicing, RNA-seq offers the opportunity to detect disease-associated splicing events, even with challenges such as the potential confounding effects of gene expression levels. The fine resolution provided by RNAseq makes it possible to visualize the global dysregulation of splicing and detect mutations in splicing-regulated genes such as SF3B1. Such observations have been documented in a large number of tumors, highlighting the relevance of RNA-seq in uncovering the complex landscape of alternative splicing. As RNA-seq has become more popular for transcriptome studies, various computational tools have been developed to address the challenges of alternative splicing analysis.
Computational tools for quantifying isoforms and alternative splicing by RNA-seq:
- Reference-based approaches: Tools such as Cufflinks and StringTie align reads to known reference genomes and leverage annotations to assemble and quantify transcript isoforms.
- Alignment-free methods: Programs such as Kallisto and Salmon utilize k-mer-based algorithms to quickly estimate transcript abundance without the need for a full sequence alignment.
- Exon-centric analysis tools: Software such as MISO and SUPPA can directly quantify exon inclusion or alternative splicing events without the need to reconstruct full-length isoforms.
- De novo assembly: For organisms without high-quality reference genomes, tools such as Trinity provide de novo transcriptome assembly, enabling researchers to study alternative splicing in non-model organisms.
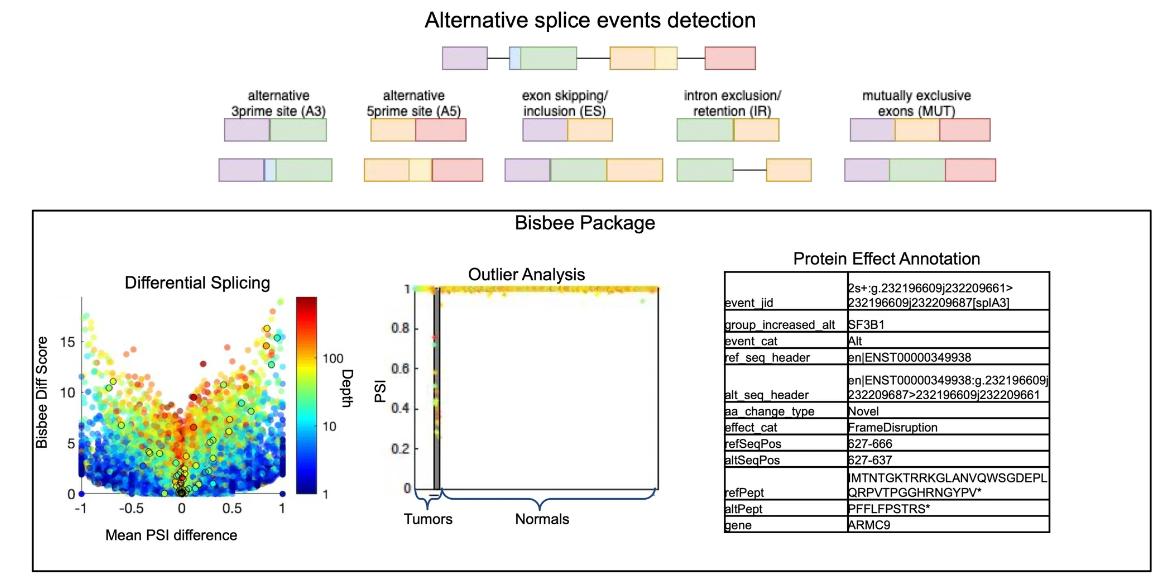
Improved methods for RNAseq-based alternative splicing analysis. (Jiang, et al., 2021)
Long-read sequencing leverages technologies such as Pacific Biosciences and Oxford Nanopore to generate longer reads than second-generation sequencing platforms. These long reads inherently facilitate the identification of genotypes without the need to reconstruct transcript variants. In the absence of high-quality reference genomes, long-read sequencing has come to the rescue, providing deeper insights into alternative splicing patterns.
A major advantage of long-read sequencing is the ability to sequence full-length RNA molecules, allowing complete isoforms to be identified without computational assembly. This capability allows for the unambiguous characterization of isoforms in a single dataset, making long-read sequencing especially important in the study of diseases in which aberrant splicing plays a key role.
IsoSplitter and Longcell: New Software Tools for Alternative Splicing
Long-read sequencing aims to sequence full-length RNA molecules, facilitating the identification of alternatively spliced isoforms. However, in the absence of a reference genome, accurate mapping of splice sites is difficult due to the diversity of alternative splicing (AS) patterns. A versatile tool, IsoSplitter, was developed based on long-read transcriptome data to backtrack and validate alternatively spliced gene "split sites". This versatile tool utilizes a modified SIM4 program to detect transcript-splitting sites, which are then quantified to reveal transcript diversity. By grouping potential isoforms, IsoSplitter provides a nuanced look at the complexity of the transcriptome, which is valuable for research efforts focused on alternative splicing in model and non-model organisms. The cornerstone of IsoSplitter is its ability to identify and characterize alternative splicing without relying on a reference genome. This feature sets it apart from many tools in the field, making it an indispensable tool for in-depth splicing analysis in non-model organisms.
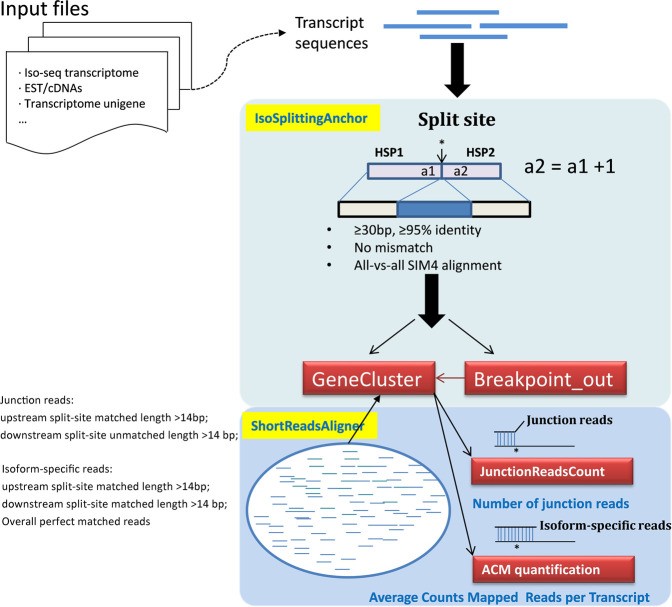
A schematic diagram of IsoSplitter design. (Wang et al., 2021)
Long-read sequencing has emerged as a powerful tool for alternative splicing analysis. However, higher sequencing errors of long reads, especially high indel rates, limit the accuracy of cellular barcodes and unique molecular identifier (UMI) recovery. Read truncation and mapping errors (the latter exacerbated by higher sequencing error rates) can lead to false detection of spurious novel isoforms. Downstream, there is not yet a rigorous statistical framework for quantifying splicing variation within and between cells/spots. Longcell is a statistical framework and computational pipeline developed for precise isoform quantification of single-cell and spatially point-barcoded long-read sequencing data. It is tailored for barcoded long-read sequencing data. By addressing the challenges of cellular barcode and unique molecular identifier (UMI) recovery, Longcell stands out for its ability to handle read truncation and mapping errors. One of the distinguishing features of Longcell is its ability to rigorously quantify the level of diversity in exon usage between cells or spots. This precision allows researchers to reveal the intricate details of intracellular splicing heterogeneity, revealing the coexistence of multiple isoforms within a single cell.
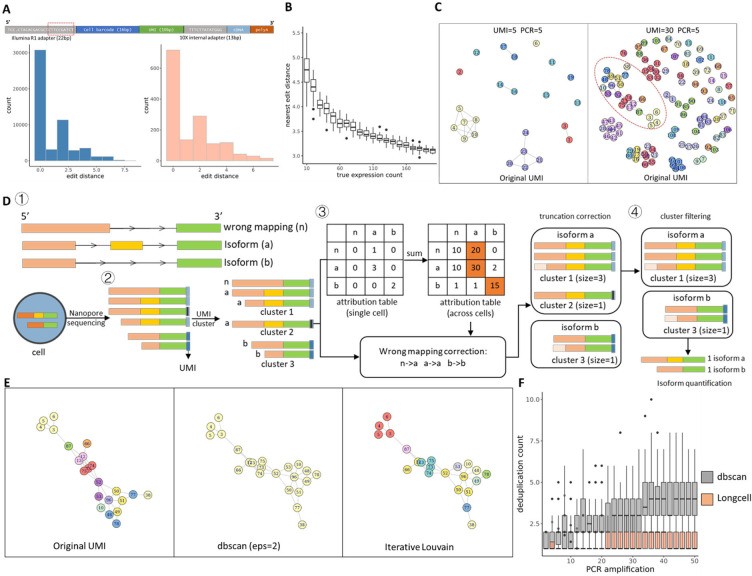
Overview of single cell Nanopore RNA seq preprocessing. (Zhang et al., 2023)
References
- Jiang, Wei, and Liang Chen. "Alternative splicing: Human disease and quantitative analysis from high-throughput sequencing." Computational and structural biotechnology journal 19 (2021): 183-195.
- Halperin, R. F., et al. "Improved methods for RNAseq-based alternative splicing analysis. Sci. Rep. 11, 10740." (2021).
- Wang, Yupeng, et al. "IsoSplitter: identification and characterization of alternative splicing sites without a reference genome." RNA 27.8 (2021): 868-875.
- Zhang, Nancy, et al. "Single cell and spatial alternative splicing analysis with long read sequencing." (2023).
Comments
Please login above to comment.